


一、CE标志制度
CE标志制度是欧盟对产品进入欧盟市场进行的监管方式。加贴CE标志的产品表明产品符合欧盟有关安全、健康、环保等法规要求,可以在欧盟27个成员国、欧洲贸易自由区的4个国家、以及英国和土耳其合法上市销售。按照欧盟规定,不同产品采用不同的评价方式加贴CE标志,主要有两种方式:绝大部分产品是制造商采取自我符合性声明方式,就可以加贴CE标志;部分风险相对更高的产品需要经过欧盟授权的第三方机构,即公告机构(Notified Body)进行符合性评定后,方可加贴CE标志。
欧盟针对不同的产品制定了不同的法规(指令),比如儿童玩具、低电压电器、个人防护器具、医疗器械等都有对应的法规(指令)。欧盟法规(指令)规定了对应产品的质量安全基本要求,以及上市的流程和合格评定程序。
产品经制造商自我声明加贴CE标志的流程:制造商必须确保自己的产品符合欧盟法规,产品是安全有效的,并建立相关技术文件,签署符合性声明,到成员国主管当局注册登记之后,即可在产品加贴CE标记进入欧盟销售。
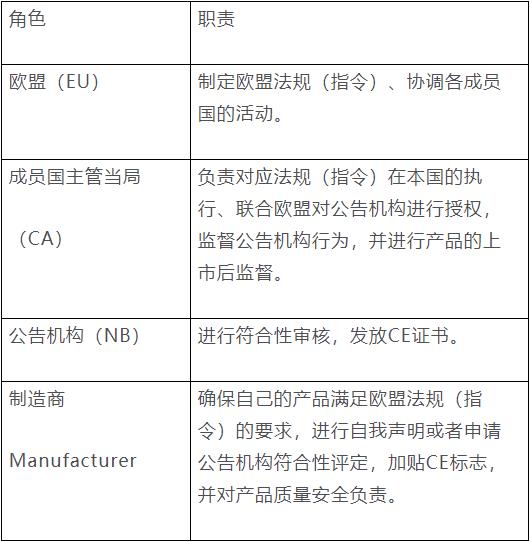
产品经公告机构符合性评定后加贴CE标志的流程:产品制造商向公告机构提出申请,公告机构为制造商提供符合性评定服务,制造商及产品符合法规要求的,向制造商发放CE证书。制造商依据CE证书签署符合性声明,产品加贴CE标志后就可以进入欧盟市场。下表列举了欧盟、成员国的主管当局、公告机构和制造商的职责。
二、中欧口罩分类及相应标准
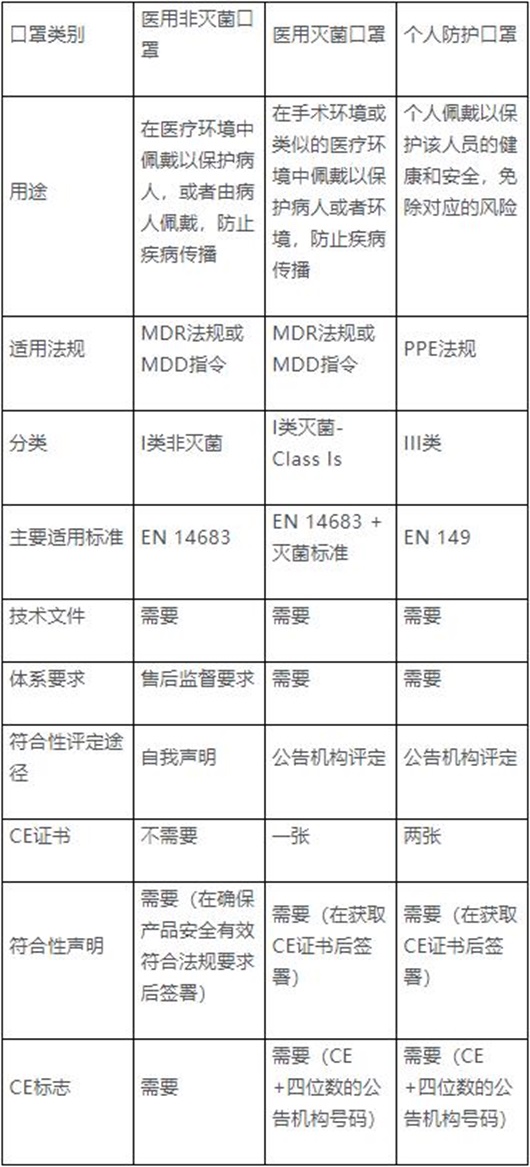
口罩在欧盟根据预期用途的不同,分为医用口罩和个人防护口罩两种,分别归属医疗器械条例EU2017/745(MDR)或医疗器械指令93/42/EEC(MDD)和个人防护设备条例EU2016/425(PPE)进行管理。如何判定具体产品属于哪一种口罩,需参照对应的法规规定和标准要求。
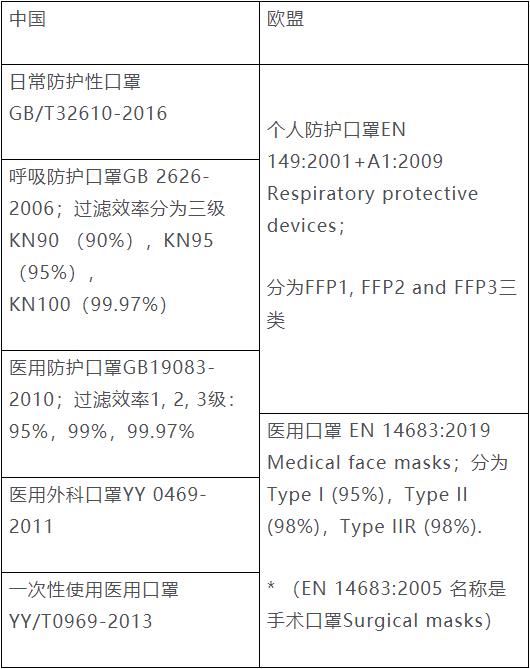
我国通常将口罩分为民用口罩和医用口罩。为便于理解,特将中国口罩类型及标准和欧盟的口罩类型及适用标准相对应,帮助大家了解您的口罩可能对应的是那种欧盟口罩,以及需要满足那个标准的要求。需要说明的是这种对应并不是严格的对应,并且中欧标准要求也有差异,请大家自己根据标准的详细要求分析并判定。
三、口罩出口欧盟的法规要求
(一)医用口罩
按照MDD或者MDR的要求,医用非灭菌口罩可以由制造商采取自我声明方式,加贴CE标志后上市销售。而医用灭菌口罩必须经公告机构符合性评定,才能加贴CE标志上市销售。但不管是否需要公告机构符合性评定,制造商都需要按照法规要求,参照相关标准或者满足欧盟质量要求的标准进行测试,以证实产品安全有效,并建立技术文件和质量管理体系,保证产品的质量安全和持续有效。
1.技术文件要求:
参照MDR法规附录II和附录III的要求(MDD为附录7),技术文件通常包括以下七个部分:
> 器械的描述和规范,包括名称、预期用途、分类、原料、构成、技术规范等。
> 产品的标签和(或)说明书
> 产品设计和制造的相关的信息
> 满足基本安全和通用性能的要求(附录I GSPR)
> 受益和风险分析,及风险管理文档
> 产品的验证和确认,包括临床前的测试和临床(评估)数据
> 上市后监督计划
2.制造商质量管理系要求:
质量管理体系可以参照协调性标准ISO 13485:2016进行。需要说明的是:质量管理体系需要参照ISO13485:2016来运行或者审核,但对于公告机构符合性评定来说,ISO13485认证证书并不是必须或者强制的,但多数的制造商都会选择取得ISO 13485认证证书,这样可以提高客户对制造商能满足法规要求及产品质量保证的信心。
3.制造商授权欧盟代表
对于欧盟境外的制造商(如中国的制造商),需要在欧盟境内授权一个欧盟代表,代替制造商在欧盟进行相关活动,比如在主管当局进行自我声明产品的登记和不良事件的报告等。制造商需要和授权欧盟代表签订协议,并规定各自承担的职责。按照新的MDR法规,制造商需要将整套的技术文件提交给欧盟代表,以便主管当局备查。
(二)个人防护口罩
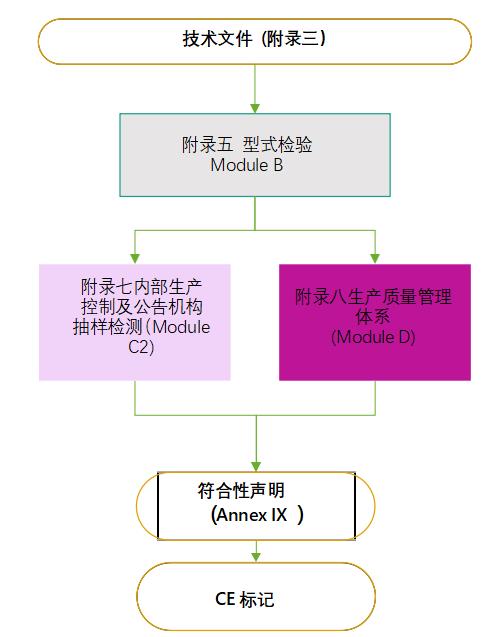
按照PPE法规要求,个人防护口罩需要有公告机构审核发放的CE证书,才能在欧盟合法上市销售。根据PPE法规关于符合性的相关规定,制造商需要建立相应技术文件,并满足EN149:2001+A1:2009的规格和测试要求。和医用口罩相比,个人防护口罩至少需获得两张公告机构的证书,即按照附录五的型式检验证书,按照附录七(Module C2)的证书或者附录八(Module D)的证书,详见下图:
四、国内具备欧盟公告机构口罩等业务资质的认证机构名录(更新至2020年4月13日)
五、国内可以开展医疗器械管理体系(ISO13485)认证的机构名录(更新至2020年4月13日)
END


